Capítulo 2 Fuentes de datos biológicas
La célula es la unidad estructural, funcional y biológica básica de todos los organismos vivos conocidos. Los organismos pueden clasificarse como unicelulares (compuestos de una sola célula, incluidas las bacterias) ó multicelulares (incluidas las plantas y los animales). Existen dos tipos de células, las eucariotas, que contienen núcleo celular, y las procariotas, que no lo contienen.
Las células eucariotas están compuestas por diversos orgánulos como la membrana, el citoplasma y el núcleo (Figura 2.1). La membrana envuelve y protege a la célula, y regula lo que entra y sale (selectivamente permeable). Dentro de la membrana, el citoplasma ocupa la mayor parte del volumen de la célula, y la separa del núcleo celular. El más prominente de los orgánulos es el núcleo celular, el cual aloja el material genético de la célula. El material genético se presenta como Acido DesoxirriboNucleico (ADN), el cual está organizado en una o más moléculas, llamadas cromosomas.
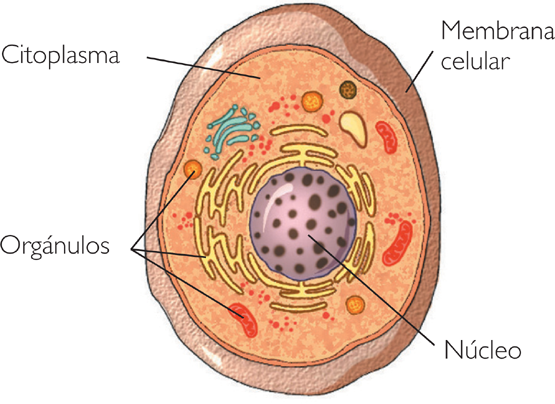
Figure 2.1: Diagrama simplificado de una célula eucariota. Imagen extraída de https://biologiarubenurjc.wordpress.com/2012/03/19/membrana-nucleo-y-citoplasma/.
La información biológica contenida en un organismo está codificada en su secuencia de ADN. El ácido desoxirribonucleico esta organizado en dos cadenas que se enrollan una alrededor de la otra para formar una doble hélice (Figura 2.2) que lleva las instrucciones genéticas utilizadas en el crecimiento, desarrollo, funcionamiento y reproducción de todos los organismos conocidos. El ADN esta conformado por unidades más pequeñas conocidas como nucleótidos. Cada nucleótido está compuesto por un azúcar llamado desoxirribosa, un grupo de fosfatos, y por una de cuatro bases nitrogenadas que son la citosina (C), guanina (G), adenina (A) ó timina (T). Los nucleótidos están unidos entre sí en una cadena por enlaces covalentes entre el azúcar de un nucleótido y el fosfato del siguiente. Las bases nitrogenadas de las dos cadenas de nucleótidos separadas se unen, según las reglas de emparejamiento de bases (A con T y C con G). Ambas cadenas de ADN almacenan la misma información biológica. Las regiones relevantes del ADN se encuentran localizadas en los cromosomas y se denominan genes. Si bien la cadena de ADN contiene millones de nucleótidos, solo un pequeño porcentaje de ella codifica proteínas (alrededor del 2% para los humanos).
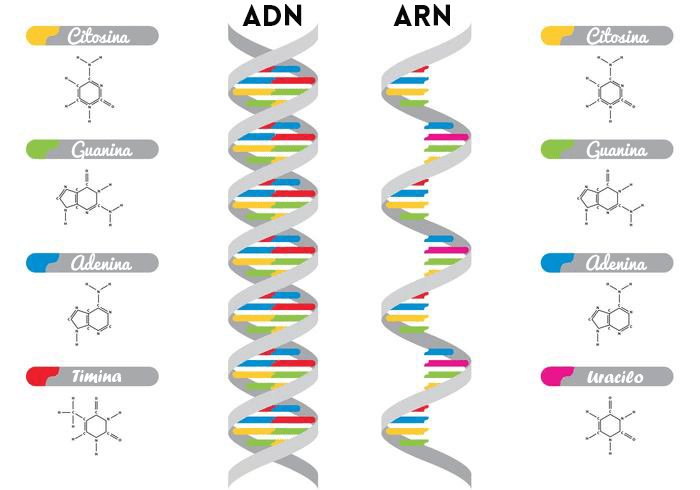
Figure 2.2: Diagrama de la estructura del ADN y ARN. Imagen extraída de https://diferencias-entre.org/diferencias-entre-adn-y-arn/.
Las células utilizan el ADN para el almacenamiento de información a largo plazo. Por otra parte, para las demás tareas en que sea necesaria la información genética, las células utilizan el Ácido RiboNucleico (ARN). El ARN se obtiene a partir del ADN para llevar a cabo tareas celulares como la síntesis de proteínas, las cuales son cadenas de aminoácidos que tienen funcionalidades básicas tanto para el metabolismo como para la fisiología celular y, en consecuencia, del organismo. La decodificación del material genético comienza dentro del núcleo celular, donde las hebras de ARN se crean utilizando el ADN como plantilla en un proceso llamado transcripción. Al igual que el ADN, el ARN se ensambla como una cadena de nucleótidos, pero a diferencia del ADN, se encuentra como una única hebra (Figura 2.2), donde las bases timina son reemplazadas por uracilo (U).
Los organismos celulares utilizan ARN mensajero (ARNm) para transmitir información genética. Orgánulos llamados ribosomas procesan el ARNm tomando cada combinación de tres nucleótidos para codificar cada uno de los 20 aminoácidos posibles. Posteriormente, los ribosomas generan la cadena de aminoácidos decodificados y así conforman la proteína codificada.
De los resultados de estos procesos celulares, existen diversos aspectos biológicos de interés científico: el genoma, el proteoma, el metaboloma, entre otros, los cuales se conocen como las diversas fuentes ómicas. A partir de una muestra biológica, para cada fuente ómica, existen tecnologías capaces de medir sus niveles de expresión. Es decir, para una misma muestra es posible obtener niveles de expresión tanto de genes, proteínas, etc.
En la presente tesis nos centraremos únicamente en aquellas ómicas que permitan obtener sus niveles de expresión en forma de matriz. Por ejemplo, en genómica, al secuenciar m muestras, es posible obtener una matriz \(G_{g*m}\), con expresión obtenida para g genes, donde \(G[i,j]\) será un valor numérico representando el nivel de expresión del i-ésimo gen para la j-ésima muestra.
2.1 Tecnologías de obtención de expresión biológica
La presente sección tiene como objetivo mencionar brevemente aspectos pertinentes sobre las tecnologías mediante las cuales se obtienen los niveles de expresión de las ómicas analizadas en la tesis.
2.1.1 Microarreglos de ADN
Un microarreglo es una superficie sólida donde pequeños fragmentos de ADN (sondas) son dispuestos en forma de matriz bidimensional. Cada celda contiene secuencias de ADN correspondientes a genes, ligados químicamente en cada celda. Para medir la expresión génica se extrae ARNm de muestras biológicas, este ARNm es luego copiado (transcripción reversa) obteniendo como resultado ADN complementario o ADNc. A este último se lo amplifica incorporando moléculas fluorescentes en las réplicas. Las copias fluorescentes del ADNc se vuelcan luego, sobre el microarreglo. De esta manera, las secuencias marcadas con moléculas fluorescentes se hibridizan (se “pegan”) a su cadena complementaria presente en el microarreglo. Luego, el microarreglo se escanea excitando las celdas con un láser y midiendo la intensidad de luz emitida por las moléculas fluorescentes. El resultado de escanear un microarreglo es una imagen por cada microarreglo. La intensidad medida en cada celda es, en principio, proporcional a la cantidad de ARNm, específico para esa celda, presente en la muestra biológica (Fernández, Alvarez, Podhajcer, & Stolovitzky, 2007).
Las imágenes resultantes del escaneo son procesadas por programas informáticos que identifican las celdas del microarreglo y miden la intensidad de luz registrada. Como resultado del procesamiento de la imagen se obtiene una serie de datos por cada celda perteneciente a cada microarreglo. En particular se obtienen los valores de intensidad de la celda, de la intensidad que rodea a la celda (intensidad de fondo), algunos índices que aportan información sobre las características de la celda (por ejemplo, el área y el perímetro de la misma) y la distribución de las intensidades dentro de cada celda. Toda esta información se utiliza para determinar la calidad del escaneo (Fresno et al., 2014). Una vez eliminadas aquellas celdas defectuosas o que presentan niveles de calidad de señal inadecuados, se normalizan los valores, comúnmente aplicando logaritmo. Finalmente se obtiene como resultado un archivo con la intensidad o el nivel de expresión de cada celda o gen.
Luego de procesar los archivos resultantes de microarreglos, y repetir el procedimiento para varios sujetos, se llega a una matriz de expresión con genes en filas y sujetos en columnas. Cada valor de la matriz representa la intensidad o nivel de expresión de un gen para una muestra. Luego de una correcta normalización de esta matriz, se llega a una del estilo a la que se muestra a continuación; sub-matriz de \(6\times5\) de datos reales provenientes de microarreglos de ADN:
A2-A0CM-01A A2-A0D0-01A A2-A0D1-01A A2-A0D2-01A A2-A0EQ-01A
ZBTB16 -0.12125000 0.00425 -0.98975 3.31300000 0.5387500
DNAJB13 -0.48200000 -0.53250 -0.60450 -0.61750000 -0.2670000
SFRP5 -0.02033333 0.22900 0.28440 -0.03216667 -0.3128333
RRAGC 1.23525000 0.68625 0.89750 1.71275000 0.9832500
IAPP 0.11500000 0.06250 1.26450 0.43500000 1.0490000
ELMO1 0.19487500 -0.35275 -0.66825 0.36437500 0.6860000Dado que los valores de expresión provienen de niveles de intensidad de luz, los datos son valores continuos. Por ende, es de esperar que la distribución de los genes, para cada sujeto, se asemeje a una distribución \(Normal\), como se observa en la Figura 2.3.
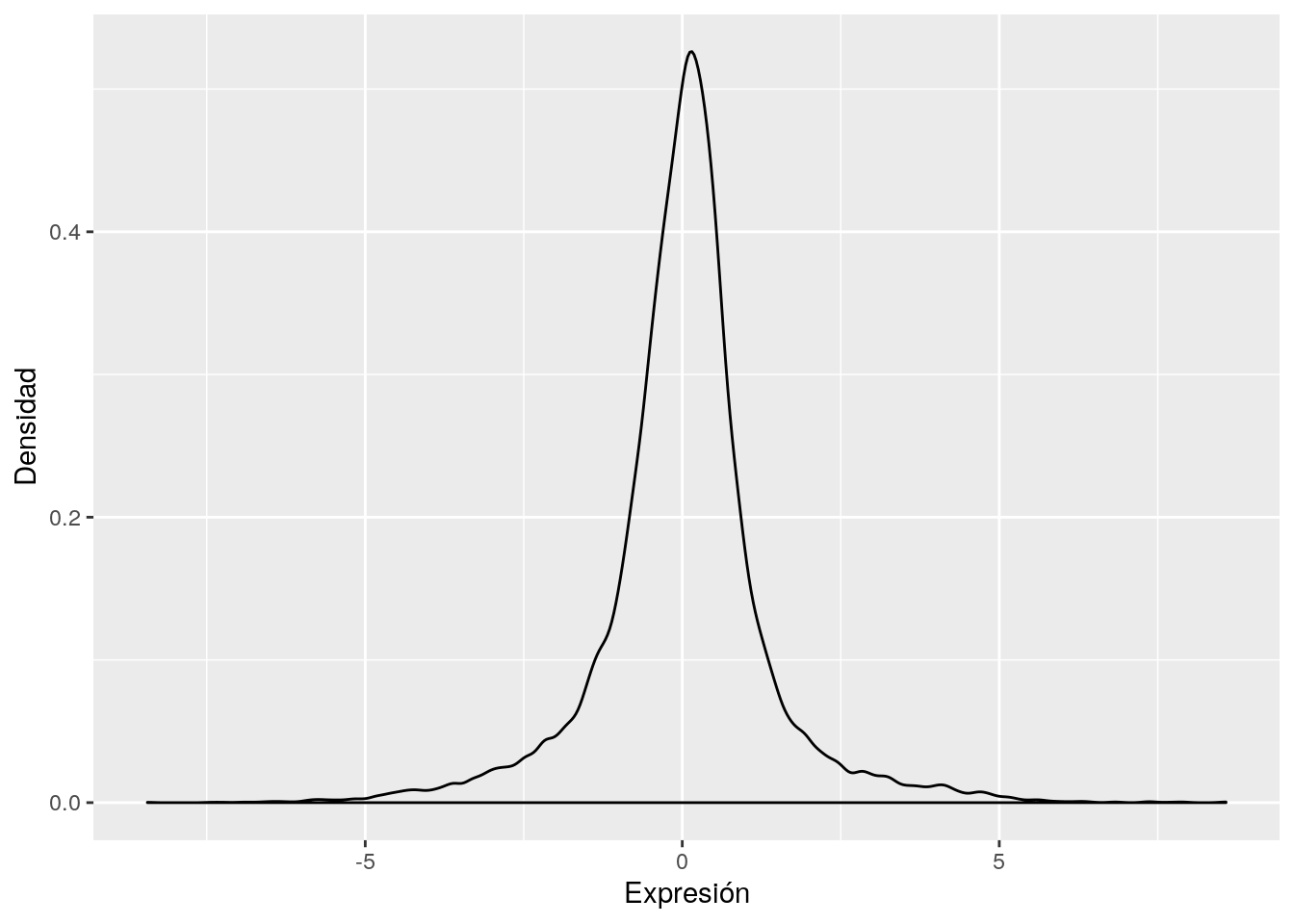
Figure 2.3: Densidad de los valores de expresión de los genes de un sujeto, para datos obtenidos mediante Microarreglos de ADN.
2.1.2 iTRAQ
El método iTRAQ se basa en el marcado químico, con etiquetas de masa variable, de las aminas de los péptidos de las digestiones de proteínas presentes en una muestra biológica. Actualmente hay dos reactivos utilizados principalmente, que pueden usarse para marcar todos los péptidos de diferentes muestras. Estas muestras luego se agrupan y generalmente se fraccionan mediante cromatografía líquida, y se analizan mediante espectrometría de masas en tándem. Luego se realiza una búsqueda en la base de datos utilizando los datos de fragmentación para identificar los péptidos marcados y, por lo tanto, las proteínas correspondientes. La fragmentación de la etiqueta adjunta genera un ión indicador de baja masa molecular que se puede usar para cuantificar relativamente los péptidos y las proteínas a partir de las cuales se originaron.A nivel peptídico, las señales de los iones indicadores de cada espectro permiten calcular la abundancia relativa (ratio) de los péptidos identificados por este espectro. Las proporciones combinadas de los péptidos de una proteína representan la cuantificación relativa de esa proteína. De esta manera se obtiene una matriz resultante de proteínas \(\times\) sujetos, con valores del ratio de la expresión de una proteína para un sujeto dado. Si bien la matriz esta indexada a nivel de proteínas, resulta de mayor interés estudiarla a nivel de genes, por ello, consultando bases de datos de anotación, se traduce cada proteína al gen que la produce. Luego de una normalización adecuada se obtiene una matriz como la que se presenta:
A2-A0CM-01A A2-A0D0-01A A2-A0D1-01A A2-A0D2-01A A2-A0EQ-01A
RRAGC 0.17794796 0.27751403 -0.13956920 0.098823426 -0.1745463
ELMO1 0.39888804 0.30589111 -0.09178467 -0.126851824 0.7252130
BAX 0.35139262 0.08414816 0.23821506 -0.118018092 -0.1782356
PDCD4 0.04627689 0.02277544 0.65416083 -0.353588366 -0.3033616
PDCD2 -0.19497549 0.55829865 -0.15272404 -0.024984301 0.6028903
PTPN6 0.17697246 0.19056327 -0.38121552 -0.002048376 0.8563106Dado que los valores de expresión provienen de niveles de señales, los datos son valores continuos. Por ende, es de esperar que la distribución se asemeje a una distribución \(Normal\), como se observa en la Figura 2.4. Vale la pena aclarar que ya que esta matriz sigue una distribución similar a la obtenida mendiante Microarreglos de ADN, es común que se utilicen los mismos métodos de análisis para ambas fuentes de datos.
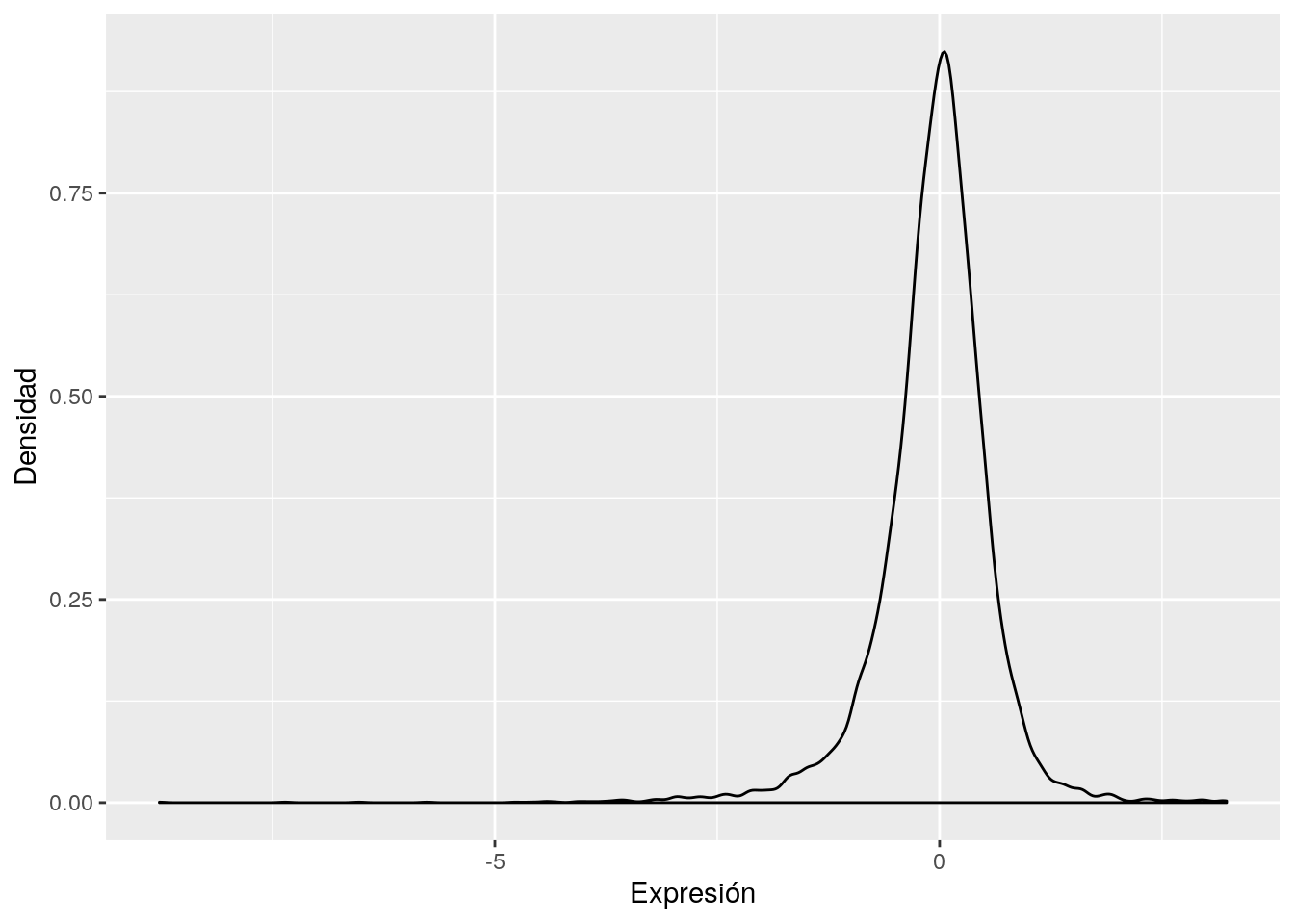
Figure 2.4: Densidad de los valores de expresión de los genes de un sujeto, para datos obtenidos mediante iTRAQ.
2.1.3 Secuenciación de ARN
Este tipo de tecnologías se basa en poder obtener para una muestra biológica, las secuencias de nucleótidos detectadas. En este sentido se obtienen millones de cadenas de nucleótidos, que al mapearlos (unirlos) permiten obtener la cantidad de veces que subcadenas del genoma aparecen en la muestra. Al poseer información a nivel de nucleótidos, surgen ventajas con respecto a los microarreglos de ADN, por ejemplo, poder detectar mutaciones de nucleótidos, e incluso estudiar la muestra a niveles más detallados que genes (transcriptos, exones, intrones, etc.).Una vez obtenido el ARN de la muestra biológica, se retrotranscribe para obtener ADN complementario (ADNc) a estas cadenas. El proceso siguiente es la fragmentación en donde, mediante cortes en secciones aleatorias, se llevan estas grandes cadenas de ADNc a fragmentos de entre 300 y 1000 nucleótidos. Cada uno de estos fragmentos es posteriormente clonado varias veces, produciendo millones de copias de cada fragmento. Aquí es cuando comienza el proceso de secuenciación propiamente dicho. En el caso de la secuenciación por síntesis, por ejemplo de las plataformas de Illumina, los fragmentos de ADNc se ponen en un pool que contiene nucleótidos individuales marcados con fluoróforos, cada una de las cuatro bases con un color diferente. Al entrar en contacto el ADNc con estos nucleótidos libres, se incorporan como hebra complementaria del ADNc, de esta manera se logra incorporar un color a cada una de las bases de los fragmentos de ADNc. Luego, cada fragmento pasa por un sistema que permite leer el color incorporado a cada nucleótido. Es así que traduciendo los colores, se logra detectar los nucleótidos que componen cada fragmento. Al finalizar el proceso de secuenciación de ARN, se cuenta con un archivo con millones de lecturas, una por cada fragmento. Cada lectura se representa con 4 líneas en el archivo, como la que se muestra a continuación:
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65Al momento inicial, cuando se obtiene el ARN de la muestra biológica, un gen más activo se encontrará más expresado que el resto, y por consiguiente, el gen presentará mayor cantidad de fragmentos asociados. Para obtener el nivel de expresión de cada gen ó transcripto particular, simplemente se puede tomar la cantidad de fragmentos que fueron mapeados al mismo. De este modo, mediante secuenciación de ARN se permite obtener una matriz de genes \(\times\) sujetos, con un conteo para cada gen y sujeto, como se puede apreciar a continuación:
A2-A0CM-01A A2-A0D0-01A A2-A0D1-01A A2-A0D2-01A A2-A0EQ-01A
ZBTB16 89 31 126 449 81
DNAJB13 11 11 44 31 43
SFRP5 1 2 0 0 0
RRAGC 2797 1453 1680 4573 2631
IAPP 162 78 167 151 195
ELMO1 1740 677 540 1758 4204Dado que los valores de expresión se desprenden de la cantidad de fragmentos mapeados, los datos son valores de conteos (no continuos). Por ende, es de esperar que la distribución de los genes, para cada sujeto, se asemeje a una distribución de \(Poisson\) o \(Binomial Negativa\), como se observa en la Figura 2.5.
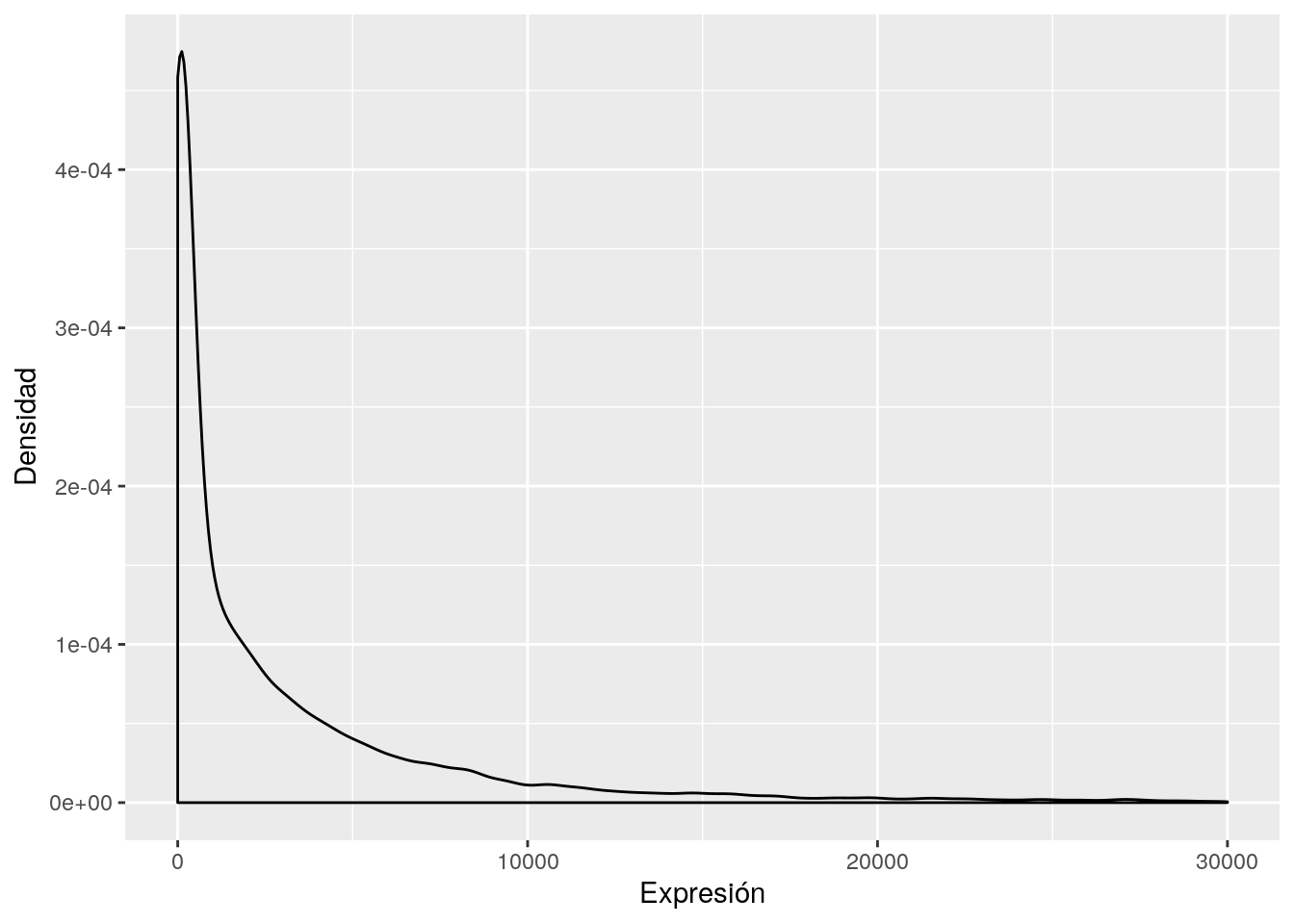
Figure 2.5: Densidad de los valores de expresión de los genes de un sujeto, para datos obtenidos mediante Secuenciación de ARN.
Con el desarrollo de este tipo de tecnología no solo se logró obtener información de expresión a nivel de genes, sino que al tener las cadenas de nucleótidos, es posible extraer información de expresión a niveles biológicos menores que genes, como ser transcriptos, exones, isoformas, etc.
2.2 Repositorios de datos de expresión
Para los diversos análisis realizados en la presente tesis, nos centramos principalmente en datos de cáncer de mama. Sin embargo, también se utilizaron repositorios con datos de cáncer de próstata. Un detalle de las bases de datos de libre acceso utilizadas se puede observar en la Tabla ??. A lo largo de este trabajo se utilizaron un total de 25 matrices de expresión de cáncer de mama y cuatro de cáncer de próstata, provenientes de microarreglos de ADN.
Adicionalmente se utilizaron datos de cáncer de mama provenientes del proyecto “el atlas del genoma del cáncer” (TCGA; del inglés The Cancer Genome Atlas). El proyecto TCGA provee, para una misma muestra, niveles de expresión provenientes de diversas fuentes ómicas, de este proyecto se utilizaron matrices de expresión de microarreglos de ADN, de proteínas medidas mediante iTRAQ, y de genes medidos mediante secuenciación de ARN. Vale la pena aclarar que no necesariamente para toda muestra perteneciente al TCGA se cuenta con datos provenientes de las tres tecnologías, en este sentido, se cuenta con 97 sujetos con muestras de las tres tecnologías.
| Nombre | Cáncer | Tecnología | #Genes | #Muestras | Referencias |
|---|---|---|---|---|---|
| Camcap | Próstata | Microarreglos | 18.718 | 199 | (Ross-Adams et al., 2015) |
| Grasso | 17.289 | 122 | (Grasso et al., 2012) | ||
| Taylor | 17.950 | 179 | (Taylor et al., 2010) | ||
| Varambally | 17.043 | 19 | (Varambally et al., 2005) | ||
| Mainz | Mama | 13.091 | 200 | (Schmidt et al., 2008) | |
| Nki | 13.120 | 337 | (Van’t Veer et al., 2002) | ||
| Transbig | 13.091 | 198 | (Chin et al., 2006) | ||
| Unt | 18.528 | 133 | (Sotiriou et al., 2006) | ||
| Upp | 18.528 | 251 | (Miller et al., 2005) | ||
| Vdx | 13.091 | 344 | (Minn et al., 2007) | ||
| (Wang et al., 2005) | |||||
| Cal | 13.091 | 118 | (Chin et al., 2006) | ||
| Dfhcc | 20.365 | 115 | (Li et al., 2010) | ||
| Dfhcc2 | 20.365 | 84 | (Silver et al., 2010) | ||
| Dfhcc3 | 20.365 | 40 | (Richardson et al., 2006) | ||
| Duke2 | 20.389 | 160 | (Bonnefoi et al., 2007) | ||
| Emc2 | 20.365 | 204 | (Bos et al., 2009) | ||
| Eortc10994 | 13.091 | 49 | (Farmer et al., 2005) | ||
| Expo | 20.365 | 353 | (Bittner, 2005) | ||
| Hlp | 19.985 | 53 | (Natrajan et al., 2010) | ||
| Irb | 20.365 | 129 | (Lu et al., 2008) | ||
| Lund2 | 12.288 | 105 | (Saal et al., 2007) | ||
| Maqc2 | 13.091 | 230 | (Shi et al., 2006) | ||
| Mccc | 19.949 | 75 | (Waddell et al., 2010) | ||
| Mda4 | 13.091 | 129 | (Liedtke et al., 2008) | ||
| (Hess et al., 2006) | |||||
| Msk | 13.091 | 99 | (Minn et al., 2005) | ||
| Nccs | 13.091 | 183 | (Yu et al., 2008) | ||
| Pnc | 20.365 | 92 | (Dedeurwaerder et al., 2011) | ||
| Stk | 18.528 | 159 | (Pawitan et al., 2005) | ||
| Unc4 | 17.779 | 305 | (Prat et al., 2010) | ||
| TCGA | 16.207 | 547 | (Weinstein et al., 2013) | ||
| TCGA | ARN | 19.948 | 547 | (Weinstein et al., 2013) | |
| TCGA | iTRAQ | 10.625 | 105 | (Weinstein et al., 2013) |
2.3 Condiciones experimentales
Como se mencionó en la Sección 1.2, para llevar a cabo el AF, es necesario contar con dos condiciones de interés a contrastar. Es decir, dos condiciones para las cuales encontrar aquellos mecanismos biológicos que las diferencian.
2.3.1 Cáncer de mama
Para cáncer de mama, Perou et al. (Parker et al., 2009) desarrolló un clasificador - PAM50 - que, a partir de datos de microarreglos de ADN, asigna a cada sujeto en uno de 5 subtipos: Luminal A, Luminal B, Her2, Basal ó Normal. Dicha clasificación se basa en los niveles de expresión detectados, para cada sujeto, en 50 genes específicos (Parker et al., 2009). Contrastar de a pares estos grupos resulta de un alto interés biológico ya que se sabe que cada grupo es diferente al resto en aspectos como tiempo de sobrevida, reacción a distintas drogas, entre otros. Y por ende, los términos biológicos característicos de cada grupo PAM50 son los que determinan su comportamiento. Para cada sujeto se obtuvo su clasificación PAM50 mediante el paquete de R genefu (Gendoo et al., 2015).
2.3.2 Cáncer de próstata
En el caso del cáncer de próstata, ya que contamos con solo cuatro bases de datos, y una de ellas con solo 19 sujetos, no se contrastaron subtipos de la enfermedad. Para este tipo de cáncer se contrastaron aquellas muestras provenientes de tejido tumoral contra provenientes de tejido normal. De aquí se desea detectar aquellos términos biológicos que caractericen el desarrollo de un tumor maligno en comparación a uno benigno.
2.4 Comentarios finales
Gracias al rápido avance de las tecnologías de obtención de expresión biológica disminuyeron notablemente sus costos, y por ende, el aumento de proyectos internacionales con mayor número de muestras y nivel de detalle. La disponibilidad libre de estas fuentes de información biológica crearon oportunidades sin precedentes para estudiar enfermedades humanas. Habiendo grandes cantidades de bases de datos biológicas de diversas poblaciones, como de distintas tecnologías ómicas, la integración de información resulta en una herramienta clave para el estudio de enfermedades (Cleveland, 2001). Sin embargo, este tipo de estudios se viene realizando a nivel de poblaciones individuales.
Resulta fundamental llevar este tipo de análisis a la comparación de diversas poblaciones. Integrando información funcional de diversos repositorios como de distintas fuentes moleculares es posible llegar a una caracterización de cada grupo de interés. Atacando aspectos funcionales activos por uno u otro grupo bajo estudio se logra el desarrollo de terapias personalizadas. Es por ello que resulta fundamental poder realizar una comparación y caracterización de múltiples fuentes de datos y poblaciones a nivel funcional.